
Abstract
Phylogenetic systematics (e.g., cladistics) is one of the most important analytical frameworks of modern Biology. It seems to be common knowledge that within phylogenetics, ‘groups’ must be defined based solely on the synapomorphies or on the “derived” characters that unite two or more taxa in a clade or monophyletic group. Thus, the idea of synapomorphy seems to be of fundamental influence and importance. Here I will show that the most common and straightforward understanding of synapomorphy as a shared derived character is not sufficient and eventually must be rejected in favor of Nelson’s relational interpretation of such term. Arguing for this point and using three examples from previously published Apes’ genomic matrices, I explicitly demonstrate that the relationship (Pongo (Gorilla (Homo, Pan))) with Hylobatidae as a sister taxon, may be successfully recovered by three-taxon statement analysis (3TA) and three-taxon statement average consensus analysis (3TS-ACA) even if all of the evident standard shared derived molecular characters of the relationship (Pongo (Gorilla (Homo, Pan))) with Hylobatidae as a sister taxon, have been excluded from the molecular alignments. Neither conventional Maximum Parsimony nor Maximum Likelihood or Bayesian Inference can do this in such situation. Thus, our results show that the relationship (Pongo (Gorilla (Homo, Pan))) with Hylobatidae as a sister taxon has appeared, in some way, behind standard shared derived characters: the last ones could be excluded, but the relationship remains the same.

Similar content being viewed by others


References
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55:539–552
Creevey C (2004) Clan: construction of supertrees and exploration of phylogenomic information from partially overlapping datasets (version 3.0). The lab. of J McInerney, Manchester
Creevey CJ, McInerney JO (2009) Trees from trees: construction of phylogenetic supertrees using Clann. In: Posada D (ed) Springer protocols: methods in molecular biology. Bioinformatics for DNA Sequence Analysis, vol 537. Humana Press, Totowa, pp 139–161
Farris JS (1983) The logical basis of phylogenetic analysis. In: Platnick NI, Funk V (eds.) Advances in cladistics, 2. Proceedings of the 2nd meeting of the Willi Hennig society; Ann Arbor, Mich., USA, Oct. 1–4; 1981. Columbia University Press, New York, pp. 7–36
Felsenstein J (1973) Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters. Syst Zool 22:240–249
Felsenstein J (2004) Inferring phylogenies. Sinauer Associates Inc, Sunderland
Finstermeier K, Zinner D, Brameier M, Meyer M, Kreuz E, Hofreiter M, Roos C (2013) A mitogenomic phylogeny of living primates. PLoS ONE 8:e69504
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) A new algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Hennig W (1966) Phylogenetic systematics. University of Illinois Press, Urbana
Kitching IJ, Forey PL, Humphries CJ, Williams DM (1998) Cladistics: the theory and practice of parsimony analysis. Oxford University Press, Oxford
Lapointe FJ, Cucumel G (1997) The average consensus procedure: combination of weighted trees containing identical or overlapping sets of taxa. Syst Biol 46:306–312
Lapointe FJ, Levasseur C (2004) Everything you always wanted to know about the average consensus, and more. In: Bininda-Emonds RP (ed) Phylogenetic supertrees: combining information to reveal the Tree of Life. Computational biology series, vol 4. Kluwer Academic Publishers, Dordrecht, pp 87–105
Lefort V, Longueville JE, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34:2422–2424
Lehtonen S, Saaksjarvi IE, Ruokolainen K, Tuomisto H (2011) Who is the closest extant cousin of humans? Total-evidence approach to hominid phylogenetics via simultaneous optimization. J Biogeogr 38:805–808
Lewis PO (2001) A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol 50:913–925
Maddison WP, Maddison DR (2018) Mesquite: a modular system for evolutionary analysis. Version 3.51. http://www.mesquiteproject.org
Marques AC (2005) Three-taxon statement analysis and its relation with primary data: Implications for cladistics and biogeography. In: Bousquets JL, Morrone JJ (eds) Regionalizacion biogeografica en Iberoamerica y topicos afines. Universidad Nacional Autonoma de Mexico, Mexico, pp 171–180
Mavrodiev EV (2016) Dealing with propositions, not with the characters: the ability of three-taxon statement analysis to recognize groups based solely on ‘reversals’, under the maximum-likelihood criteria. Aust Syst Bot 29:119–125
Mavrodiev EV, Madorsky A (2012) TAXODIUM Version 1.0: a simple way to generate uniform and fractionally weighted three-item matrices from various kinds of biological data. PLoS ONE. 7:e48813
Mavrodiev EV, Yurtseva OV (2017) “A character does not make a genus, but the genus makes the character”: three-taxon statement analysis and intuitive taxonomy. Eur J Taxon 377(7):1–7
Mavrodiev EV, Dell C, Schroder L (2017) A laid-back trip through the Hennigian Forests. Peer J. 5:e3578
Mavrodiev EV, Williams DM, Ebach MC (2019) On the typology of relations. Evol Biol 46:71–89
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic tres. In: Pirece M (ed.) Proceedings of the gateway computing environments workshop (GCE), 14 Nov. 2010, New Orleans, pp 1–8
Nelson G (1994) Homology and systematics. In: Hall BK (ed) Homology: The hierarchical basis of comparative biology. Academic Press, Harcourt Company, San-Diego, pp 101–149
Nelson G (1996) Nullius in verba. Self-published, New York
Nelson G (2011) Resemblance as evidence of ancestry. Zootaxa 2946:137–141
Nelson G, Platnick NI (1981) Systematics and biogeography: cladistics and vicariance. Columbia University Press, New York
Nelson G, Platnick NI (1991) Three-taxon statements—a more precise use of parsimony? Cladistics 7:351–366
Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MAM, Kessing B, Pontius J, Roelke M, Rumpler Y, Schneider MCP, Silva A, O’Brien SJ, Pecon-Slattery J (2011) A molecular phylogeny of living primates. PLoS Genet 7:e1001342
Rambaut A (2012) FigTree v. 1.4. Molecular evolution, phylogenetics and epidemiology. University of Edinburgh, Edinburgh
Rannala B, Yang ZH (1996) Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol 43:304–311
Rineau V, Zaraguëta-Bagils R, Laurin M (2018) Impact of errors on cladistic inference: simulation-based comparison between parsimony and three-taxon analysis. Contr Zool 87:25–40
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schuh RT, Brower AVZ (2009) Biological systematics: principles and applications, 2nd edn. Comstock Publishing Associates, Cornell University Press, Ithaca
Schwarz GE (1978) Estimating the dimension of a model. The Ann Statist 6:461–464
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sinauer Associates Inc, Sunderland
Waddell PJ, Steel MA (1997) General time-reversible distances with unequal rates across sites: mixing G and inverse Gaussian distributions with invariant sites. Mol Phylogen Evol 8:398–414
Wiley EO, Lieberman BS (2011) Phylogenetics: the theory and practice of phylogenetic systematics, 2nd edn. John Wiley and Sons Inc, Hoboken
Williams DM, Ebach MC (2005) Drowning by numbers: rereading Nelson’s “Nullius in Verba”. Bot Rev 71:415–447
Williams DM, Ebach MC (2008) Foundations of systematics and biogeography. Springer, New York
Williams DM, Ebach MC (2016) What is intuitive taxonomic practise? Syst Biol 66:637–643
Williams DM, Siebert DJ (2000) Characters, homology and three-item analysis. In: Scotland RW, Pennington TR (eds) Homology and systematics: coding characters for phylogenetic analysis. Systematics Association Special Volume (Book 58). Taylor and Francis, London, pp 183–208
Acknowledgements
Dr. David M. Williams (Natural History Museum, London, UK) and Dr. Malte C. Ebach (University of New South Wales, AU) are highly acknowledged for their comments and useful discussion. I would like to thank Prof. Pablo Goloboff (National Scientific and Technical Research Council, Buenos Aires, Argentina) for his notes on an earlier version of this manuscript. No agreement with either the methodology or the conclusions of the paper is implied on his behalf.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10441_2019_9368_MOESM1_ESM.pdf
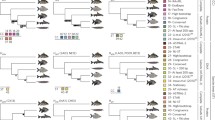
Supplementary material 1 (PDF 1159 kb). Figure S1. The results of standard MP analysis, 3TA and 3TS-ACA of the complete and reduced genomic supermatrix of the Great Apes and outgroups based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). a. Single most parsimonious tree of 12,092 steps (Consistency Index (reviewed in Kitching et al. 1998, CI hereinafter) = 0.7877; Retention Index (reviewed in Kitching et al. 1998, RI hereinafter) = 0.5597) recovered from the standard MP analysis (Fitch Parsimony) of the complete seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes and outgroups based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). The number of parsimony-informative characters is equal to 4311. Tree was a posteriori rooted relative to Macaca. b. Single nested most parsimonious hierarchy of patterns (length = 72,677; RI = 0.8435) recovered from MP analysis (Wagner parsimony) of 3TS-WS representation of the complete seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes and outgroups (a) based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). The values of the operational outgroup were fixed as values of Macaca. The number of 3TSs is equal to 62,844, all are parsimony-informative and weighted uniformly. c. Single nested hierarchy of patterns given as an average consensus of the score 0.10408 of 62,844 minimal trees (3TSs) (see b for more detail). d. Single most parsimonious tree of 5621 steps (CI = 0.9425, RI = 0.7203) recovered from the standard MP analysis (Fitch Parsimony) of the reduced seven taxa/98-loci genomic supermatrix of the Great Apes and outgroups based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017), with the total number of characters in supermatrix equal to 38920. 3302 synapomorphic characters have been excluded from the original 42,222 bp supermatrix (a). The number of parsimony-informative characters is equal to 1155. Tree was a posteriori rooted relative to Macaca. e. Single nested most parsimonious hierarchy of patterns (length = 34,040; RI = 0.9311) recovered from a MP analysis (Wagner parsimony) of 3TS-WS representation of the reduced seven taxa/38,920 characters/98-loci genomic supermatrix of the Great Apes and outgroups based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017) (d). The values of operational outgroup were fixed as values of Macaca. The number of 3TSs is equal to 31,845, all are parsimony-informative and weighted uniformly. f. Single nested hierarchy of patterns given as an average consensus of the score 0.13425 of 31,845 minimal trees (3TSs) (see e for more detail). Figure S2. The results of standard MP analysis, 3TA and 3TS-ACA of the complete and reduced mtDNA supermatrix of the Great Apes and outgroups modified from Finstermeier et al. (2013). a. Single most parsimonious tree of 10,339 steps (CI = 0.7565, RI = 0.4978) recovered from the standard MP analysis (Fitch Parsimony) of the complete seven taxa/16,563 bp mtDNA supermatrix of the Great Apes and outgroups modified from Finstermeier et al. (2013). The number of parsimony-informative characters is equal to 3602. Tree was a posteriori rooted relative to Macaca. b. Single nested most parsimonious hierarchy of patterns (length = 62,362; RI = 0.8176) recovered from a MP analysis (Wagner parsimony) of 3TS-WS representation of the complete seven taxa/16,563 bp mtDNA supermatrix of the Great Apes and outgroups (a) modified from Finstermeier et al. (2013). The values of operational outgroups were fixed as values of Macaca. The number of 3TSs is equal to 52,744, all are parsimony-informative and weighted uniformly. c. Single nested hierarchy of patterns, given as an average consensus of the score 0.07653 of 52,744 minimal trees (3TSs) (see b for more detail). d. Single most parsimonious tree of 4445 steps (CI = 0.9298, RI = 0.6605) recovered from the standard MP analysis (Fitch Parsimony) of the reduced mtDNA supermatrix of the Great Apes and outgroups (seven taxa) (a) modified from Finstermeier et al. (2013) with the total number of characters in supermatrix equal to 13835. 2728 synapomorphic characters have been excluded from the original 16,563 bp alignment (a). The number of parsimony-informative characters is equal to 3602. Tree was a posteriori rooted relative to Macaca. e. Single nested most parsimonious hierarchy of patterns (length = 28,310; RI = 0.9190) recovered from a MP analysis (Wagner parsimony) of 3TS-WS representation of the reduced seven taxa/13,835 bp mtDNA supermatrix of the Great Apes and outgroups (d) modified from Finstermeier et al. (2013). The values of operational outgroup were fixed as values of Macaca. The number of 3TSs is 26,188, all are parsimony-informative and weighted uniformly. f. Single nested hierarchy of patterns given as an average consensus of the score 0.10723 of 26,188 minimal trees (3TSs) (see e for more detail). Figure S3. The results of standard MP analysis, 3TA and 3TS-ACA of the complete and reduced genomic supermatrix of the Simiiformes and outgroups, modified from Perelman et al. (2011). a. Single most parsimonious tree of 8103 steps (CI = 0.8728, RI = 0.8585) recovered from the standard MP analysis (Fitch Parsimony) of the complete 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). All constant characters were excluded from the supermatrix before analysis. The number of parsimony-informative characters is equal to 2530. Tree was a posteriori rooted relative to Lemur. b. Single nested most parsimonious hierarchy of patterns (length = 755,427; RI = 0.9140) recovered from the standard MP analysis (Wagner parsimony) of 3TS-WS representation of the complete 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011) (a). The values of operational outgroup were fixed as values of Lemur. The number of 3TSs is equal to 686,390, all are parsimony-informative and weighted uniformly. c. Single nested hierarchy of patterns given as an average consensus of the score 0.23925 of 686,390 minimal trees (3TSs) (see b for more detail). d. Strict consensus of eight most parsimonious trees of 7740 steps (CI = 0.8829, RI = 0.8649) recovered from the standard MP analysis (Fitch Parsimony) of the reduced 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). 192 synapomorphic characters have been excluded from the original 6319 bp alignment (a). The number of parsimony-informative characters is equal to 2338. Tree was a posteriori rooted relative to Lemur. Tree was a posteriori rooted relative to Lemur. e. Single nested most parsimonious hierarchy of patterns (length = 715,673; RI = 0.9213) recovered from a MP analysis (Wagner parsimony) of 3TS-WS representation of reduced 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes modified from Perelman et al. (2011) with 192 synapomorphic characters have been excluded (d). The number of 3TSs is equal to 663,477, all are parsimony-informative and weighted uniformly. The values of operational outgroup were fixed as values of Lemur. f. Single nested hierarchy of patterns given as an average consensus of the score 0.26001 of 663,477 minimal trees (3TSs) (see e for more detail). The names of the major relationships: I. Simiiformes; II. Platyrrhini (New World Monkeys); III. Cercopithecidae; IV. Hylobatidae; V. Hominidae (Great Apes). Figure S4. The results of standard ML analysis of the complete and reduced genomic supermatrices of the Great Apes and outgroups, based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017) (a-c) and modified from Finstermeier et al. (2013) (d-f). a. Most probable tree (-log likelihood: 114,639.949003) recovered from the ML analysis of the complete seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). Numbers above branches indicate the aLRT support values. Tree was a posteriori rooted relative to Macaca. b. Most probable tree (-log likelihood: 84,393.620715) recovered from the ML analysis of the reduced seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). 3302 characters have been excluded from the original 42,222 bp supermatrix (a). Numbers above branches indicate the aLRT support values. Tree was a posteriori rooted relative to Macaca. c. The same tree as b but with all branches with aLRT support values equal to zero were collapsed. d. Most probable tree (-log likelihood: 63,763.326079) recovered from the ML analysis of the complete seven taxa/16,563 bp mtDNA supermatrix of the Great Apes, modified from Finstermeier et al. (2013). Numbers above branches indicate the aLRT support values. Tree was a posteriori rooted relative to Macaca. e. Most probable tree (-log likelihood: 39,713.360152) recovered from the ML analysis of the reduced seven taxa/13,835 bp mtDNA supermatrix of the Great Apes modified from Finstermeier et al. (2013). 2728 characters have been excluded from the original 16,563 bp supermatrix (d). Numbers above branches indicate the aLRT support values. Tree was a posteriori rooted relative to Macaca. f. The same tree as e, but with all branches with aLRT support values below 0.300 have been collapsed. Figure S5. The results of the Bayesian analysis (BI) of the complete and reduced genomic supermatrices of the Great Apes and outgroups, based on the DNA sequence data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017) (a-c) and modified from Finstermeier et al. (2013) (d-f). a. Consensus tree recovered from the BI of the complete seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.000000. Tree was a posteriori rooted relative to Macaca. b. Consensus tree recovered from the BI of the reduced seven taxa/98-loci (42,222 bp) genomic supermatrix of the Great Apes based on the data from Lehtonen et al. (2011), as summarized in Mavrodiev et al. (2017). 3302 characters have been excluded from the original 42,222 bp genomic supermatrix (a). At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.004054. Tree was a posteriori rooted relative to Macaca. c. Consensus tree recovered from a BI of the complete seven taxa/16,563 bp mtDNA supermatrix of the Great Apes modified from Finstermeier et al. (2013). At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.000000. Tree was a posteriori rooted relative to Macaca. d. Consensus tree recovered from the BI of the reduced 13,835 characters/7taxa mtDNA supermatrix of the Great Apes modified from Finstermeier et al. (2013). 2728 characters have been excluded from the original 16,563 bp genomic supermatrix (c). At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.003152. Tree was a posteriori rooted relative to Macaca. Figure S6. The results of standard ML analysis of the complete and reduced genomic supermatrix of Simiiformes and outgroups, modified from Perelman et al. (2011). a. Most probable tree (-log likelihood: 43,617.028526) recovered from the ML analysis of the complete 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). All characters are variable. Tree was a posteriori rooted relatively Lemur. Numbers above and below branches indicate the aLRT support values that have appeared to be less than 1.000. All of the other branches received aLRT support of 1.000. Tree was a posteriori rooted relatively Lemur. b. Most probable tree (-log likelihood: 41,412.621128) recovered from the ML analysis of the reduced 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). 192 characters have been excluded from the original 6319 bp supermatrix (a). All characters are variable. Tree was a posteriori rooted relative to Lemur. Numbers above and below branches indicate the aLRT support values that have appeared to be less than 1.000. All of the other branches received aLRT support of 1.000. Tree was a posteriori rooted relative to Lemur. c. The same tree as b, but with all branches with aLRT support values below 0.300. See legend of the Figure S3 for the names of the major relationships (I.-V.). Figure S7. The results of Bayesian analysis (BI) of the complete and reduced genomic supermatrix of the Simiiformes and outgroups modified from Perelman et al. (2011). a. Consensus tree recovered from a BI of the complete 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). All characters are variable. At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.000157. Tree was a posteriori rooted relative to Lemur. b. Consensus tree recovered from a BI of the reduced reduced 21 taxa/54-nuclear gene regions (6319 bp) genomic supermatrix of Simiiformes and outgroups modified from Perelman et al. (2011). All characters are variable. 192 characters have been excluded from the original 6319 bp supermatrix (a). At the end of the runs, the standard deviation of split frequencies between the two runs had fallen to 0.002332. Tree was a posteriori rooted relative to Lemur. See the legend of the Figure S3 for the names of the major relationships (I.-V.)
Rights and permissions
About this article

Cite this article
Mavrodiev, E.V. Synapomorphies Behind Shared Derived Characters: Examples from the Great Apes’ Genomic Data. Acta Biotheor 68, 357–365 (2020). https://doi.org/10.1007/s10441-019-09368-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10441-019-09368-6